

Norrish, G;
Field, E;
Mcleod, K;
Ilina, M;
Stuart, G;
Bhole, V;
Uzun, O;
... Kaski, JP; + view all
(2018)
Clinical presentation and survival of childhood hypertrophic cardiomyopathy: a retrospective study in United Kingdom.
European Heart Journal
10.1093/eurheartj/ehy798.
(In press).

Preview |
Text (Article)
ehy798.pdf - Published Version Download (458kB) | Preview |
Preview |
Text (Supplementary table 1)
Kaski_ehy798_supplementary_table_1.pdf - Accepted Version Download (129kB) | Preview |
Preview |
Text (Supplementary table 2)
Kaski_ehy798_supplementary_table_2.pdf - Accepted Version Download (136kB) | Preview |
Preview |
Text (Supplementary table 3)
Kaski_ehy798_supplementary_table_3.pdf - Accepted Version Download (24kB) | Preview |
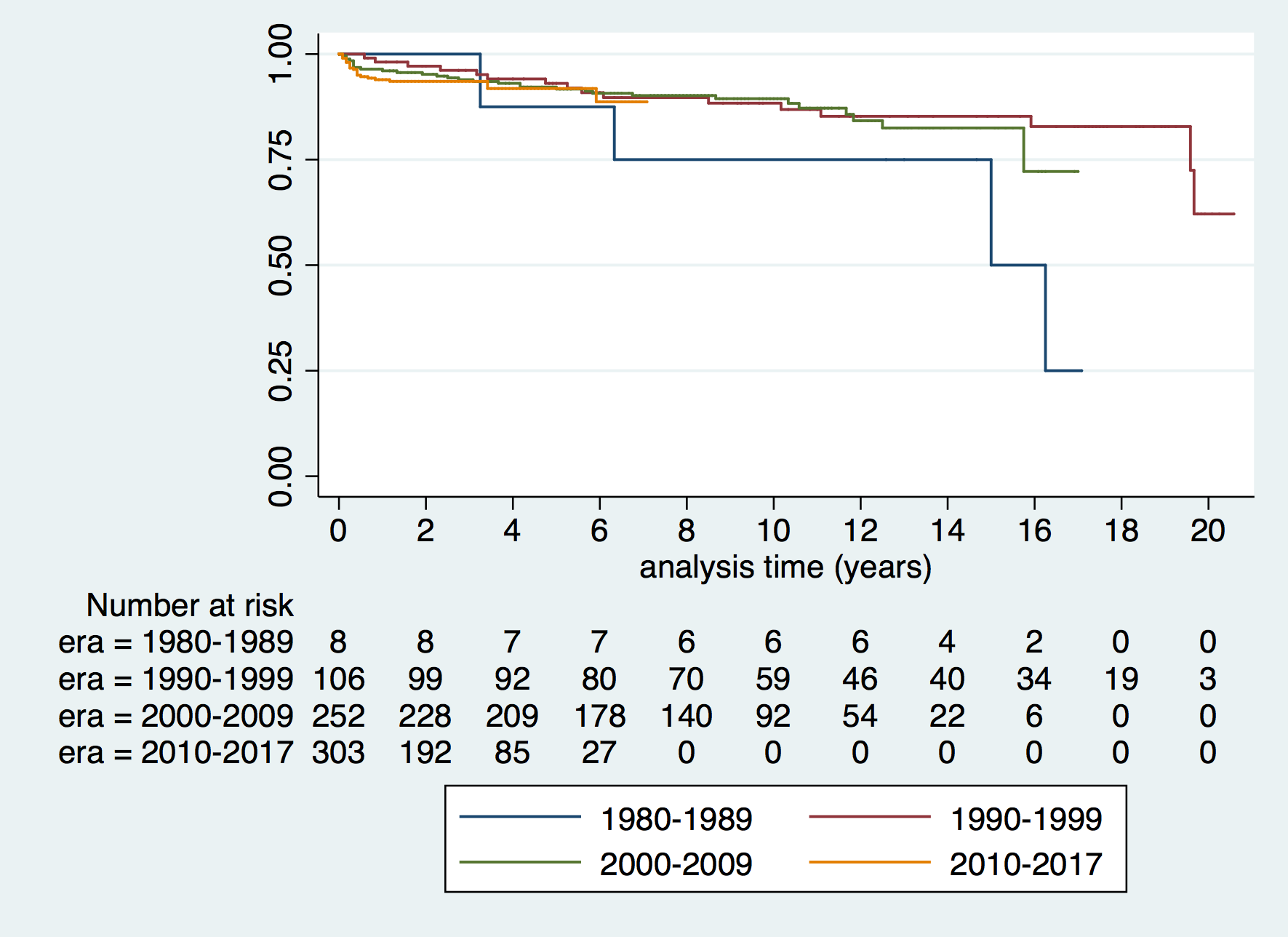
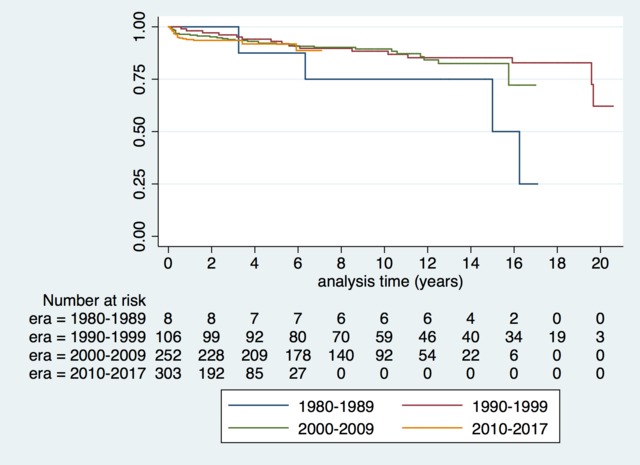
![[thumbnail of Supplementary figure 1]](https://discovery-pp.ucl.ac.uk/10065420/37/Kaski_ehy798_supplementary_figure_1.png) 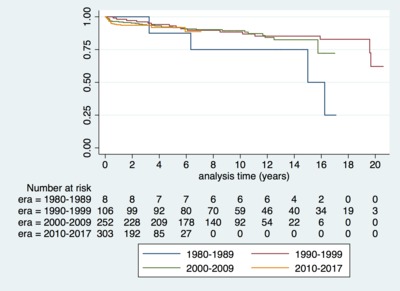 Preview |
Image (Supplementary figure 1)
Kaski_ehy798_supplementary_figure_1.png - Accepted Version Download (90kB) | Preview |
Abstract
Aims: Understanding the spectrum of disease, symptom burden and natural history are essential for the management of children with hypertrophic cardiomyopathy (HCM). The effect of changing screening practices over time has not previously been studied. This study describes the clinical characteristics and outcomes of childhood HCM over four decades in a well-characterized United Kingdom cohort. Methods and results: Six hundred and eighty-seven patients with HCM presented at a median age of 5.2 years (range 0-16). Aetiology was: non-syndromic (n = 433, 63%), RASopathy (n = 126, 18.3%), Friedreich's ataxia (n = 59, 8.6%) or inborn errors of metabolism (IEM) (n = 64, 9%). In infants (n = 159, 23%) underlying aetiology was more commonly a RASopathy (42% vs. 11.2%, P < 0.0001) or IEM (18.9% vs. 6.4% P < 0.0001). In those with familial disease, median age of presentation was higher (11 years vs. 6 years, P < 0.0001), 141 (58%) presented <12 years. Freedom from death or transplantation was 90.6% (87.9-92.7%) at 5 years (1.5 per 100 patient years) with no era effect. Mortality was most frequently sudden cardiac death (SCD) (n = 20, 2.9%). Children diagnosed during infancy or with an IEM had a worse prognosis (5-year survival 80.5% or 66.4%). Arrhythmic events occurred at a rate of 1.2 per 100 patient years and were more likely in non-syndromic patients (n = 51, 88%). Conclusion: This national study describes a heterogeneous disease whose outcomes depend on the age of presentation and aetiology. Overall mortality and SCD rates have not changed over time, but they remain higher than in adults with HCM, with events occurring in syndromic and non-syndromic patients.
| Type: | Article |
|---|---|
| Title: | Clinical presentation and survival of childhood hypertrophic cardiomyopathy: a retrospective study in United Kingdom |
| Location: | England |
| Open access status: | An open access version is available from UCL Discovery |
| DOI: | 10.1093/eurheartj/ehy798 |
| Publisher version: | http://doi.org/10.1093/eurheartj/ehy798 |
| Language: | English |
| Additional information: | Copyright © The Author(s) 2018. Published by Oxford University Press on behalf of the European Society of Cardiology. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited. |
| Keywords: | Hypertrophic cardiomyopathy, United Kingdom, Survival, Aetiology |
| UCL classification: | UCL UCL > Provost and Vice Provost Offices > School of Life and Medical Sciences UCL > Provost and Vice Provost Offices > School of Life and Medical Sciences > Faculty of Population Health Sciences > Institute of Cardiovascular Science |
| URI: | https://discovery-pp.ucl.ac.uk/id/eprint/10065420 |

{kind=link}
{kind=link}
Archive Staff Only
 |
View Item |